
Tutorial to SMILES and canonical SMILES explained with examples
My Background and Why I Wrote This Tutorial
I wrote my master thesis in chemoinformatics with a focus on drug discovery and machine learning applications. Large data bases containing various compounds in SMILES format were part of my daily work life. When I started working in chemoinformatics, I knew that SMILES referred to a formatted string that contained all the information about the graph of a molecule, however, I had no idea how it was generated on an algorithmic level. Furthermore, canonical SMILES use a labeling system to uniquely identify each compound bijectively, which is a level of complexity even above the generic SMILES format.
At a certain point it became inevitable to thoroughly understand the SMILES format and figuring out how SMILES work turned out to be quite a task. Most videos on YouTube barely scratch the surface only explaining the concept very roughly. Moreover, most articles I found so far regurgitate the two origin publications on SMILES while neither adding information nor improving on didactic aspects.
Frankly, the two origin publications illuminate a lot of concepts around SMILES but are not enough to gain a thorough understanding of the subject. Luckily, I found further sources that truly augment the origin publications. First of all daylight.com is a website, that offers educational material on the topic that is based on the origin publication but is kept up to date and contains nicer figures. All concepts necessary to understand the generic SMILES format are available from there and will be explained in this tutorial as well. The canonical SMILES generation, however, is just briefly mentioned in the daylight manual. A slide show from the University of Bonn (Germany) from Martin Vogt, features an example and an explanation that makes understanding the canonical SMILES algorithm easy.
This tutorial will enable you to understand SMILES and their canonicalization. Also I will include my sources at the end of the article to ease your citations.
Simplified Molecular Input Line Entry Specification — SMILES
SMILES refers to a specific formalism to generate identifiers for chemical compounds that are suited for chemists and computational inputs alike. The identifier, in this case, is deduced from a two-dimensional graph representing the chemical structure (skeletal formula). The result is a series of characters that contain mostly alphanumeric symbols, brackets and more. The selection of those symbols follows a specific set of rules. The set of rules addresses six categories: atoms, bonds, branches, cyclic structures, disconnected structures and aromaticity. Also, SMILES considers stereochemical information.[1]
Atoms are labelled by their element symbol. All elements of a SMILES string are written in square brackets with the exceptions of the organic subset, i.e. B, C, N, O, P, S, F, Cl, Br, and I.
Hydrogen atoms have further specifications. They can either be denoted implicitly with atoms of the organic subset. In that case, the remainder of the lowest valence is filled with hydrogen atoms. For example, C refers to CH4.
Explicit notation of hydrogen atoms occurs when they are attached to an element that is not part of the organic subset. Given a metal M, the nomenclature of four hydrogen atoms attached to that metal is [MH4]. Molecular hydrogen is denoted on its own in brackets [H]. Charges are represented with a plus or minus with their respective count inside a bracket.[1]
Within the SMILES nomenclature, bonds are omitted if they are either aromatic or single covalent bonds. Double bonds are represented with ‘=’, and triple bonds are represented by ‘#’. Ionic bonds are not explicitly denoted by the SMILES algorithm. An ion pair is written as two disconnected structures with formal charges to them.
Tautomeric bonds are not explicitly denoted either. One of the possible structures is translated into the SMILES string, be it the enol or keto variation.[1]
Branches are depicted in parenthesis. 5-Methyl-4-(2-methylpropyl)-4-(propan-2-yl)hept-1-ene is an example of nested branching using nested parentheses. The name of the structure is according to the convention of IUPAC.[2] The structure and SMILES is shown in Fig.1.

Cyclic structures are converted in-silico to linear structures by breaking a single or aromatic bond within the cycles. For SMILES generation the broken bonds are denoted by writing a number right behind the formerly connecting elements. An illustrating example with only one cycle is shown in Fig. 2.

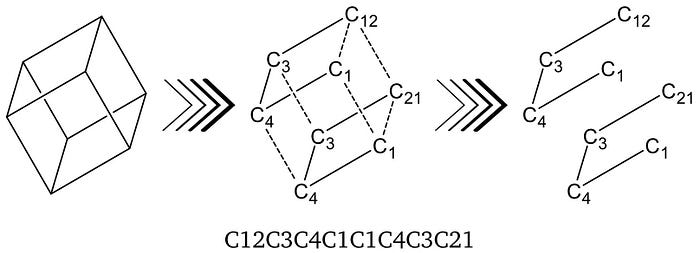
Furthermore, a single atom can be part of multiple rings which is then accounted for by using two or three single digits in sequence. However, for structures with more than ten rings, double digits are separated with a prefacing per cent sign. Also, a single digit can be reused for multiple broken bonds without creating ambiguity. A SMILES string is read from left to right, and a ring closes on the first repetition of a respective digit. Cubane has multiple rings and can illustrate the concept of multi-rings and digit-re-usability. In Fig. 3 the generation of a Cubane-SMILES string is shown with the double-usage of the digit ‘1’. [1]
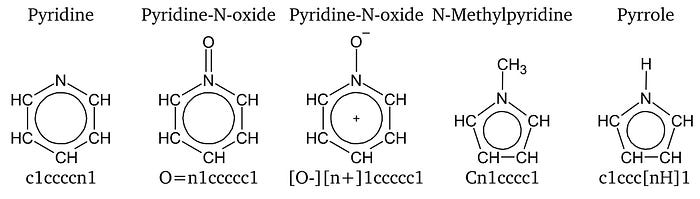
Aromaticity within SMILES is denoted by writing the atoms that are part of an aromatic cycle in lower case letters. Aromaticity is detected by applying an extended definition of Hückel’s rule. Another noteworthy convention is the treatment of aromatic nitrogen atoms. A nitrogen atom that is embraced by two aromatic bonds has no valency left per default. However, aromatic nitrogen can be connected to an additional hydrogen nonetheless. In this case, the extraordinary hydrogen atom is specified in square brackets. Structures of aromatic N-heterocycles and their SMILES are shown in Fig. 4 as examples of aforementioned rules.[1]
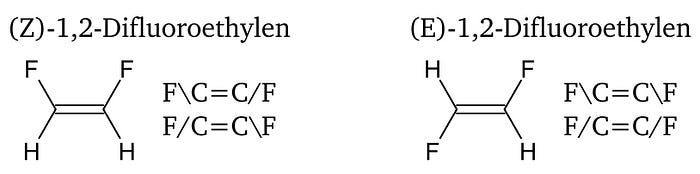
Furthermore, the SMILES algorithm introduces a convention for labeling double bond configurations and chirality. The double bond configuration is indicated by placing ‘/’ or ‘\’ between the atom constituting the double bond and their subsequent bonding partners. The indicators can be understood as a single-bond type that gives information about their relative orientation. An example for (Z)-1,2-difluoroethene and (E)-1,2-difluoroethene is given in Fig. 5.[3]
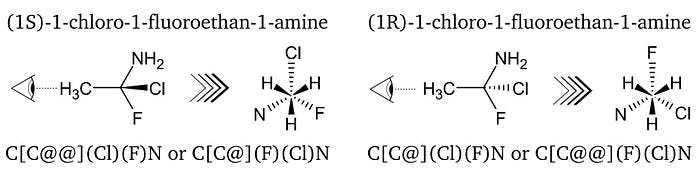
Chirality is assigned to chiral tetrahedral centres and any other chiral centre, e.g. allene-like or square planar centres. Herein, the SMILES notation is explained in the context of tetrahedral chirality centres, which is the most straightforward instance of chirality in organic chemistry. Firstly, a chiral centre can not be the terminal node in a molecular representation since a terminal node is either only connected to hydrogen atoms if any at all. With that in mind, the convention for tetrahedral chirality is most easily explained by elaborating an example, here (1S)-1-chloro-1-fluoroethan-1-amine which can be seen in Fig. 6. The whole molecule is viewed along the C-C-bond. Necessarily, the enantiomeric SMILES string contains the chiral C-atom’s binding partners in a specific order. This sequence can either correspond to the clockwise or anticlockwise order when the molecule is viewed along the C-C-bond. Should the order be anticlockwise, an ‘@’ is inserted after the central C-atom in square brackets. The ‘@’ is a visual mnemonic since it depicts an anticlockwise rotation around the central circle. For a clockwise order, ‘@@’ is used instead of a single ‘@’.
Canonical SMILES — The CANGEN Algorithm
How any SMILES for a certain structure can be generated should now be clear. The set of rules discussed above is just a matter of notation and frankly there is no deep understanding necessary. The generation of a unique SMILES representation, however, is slightly mathematical and is not as straightforward as the SMILES rule set itself.
In general, SMILES strings do not claim to be unique identifiers. There are many equivalent options to generate a SMILES string for a given structure. Nowadays, computational biochemical research accesses structures from many different sources and databases, making the requirement of a unique identifier evident. The SMILES notation was developed with this objective in mind. So-called canonical SMILES fulfil this objective. They are based on the same set of rules described in the prior section. The so-called CANGEN algorithm can be partitioned into two parts: the CANON part and the GENES part. The CANON part labels the atoms of the molecular structure canonically, i.e. a unique way based on the structural topology. The GENES part generates a unique SMILES from the canonical labeling and the aforementioned rule set (see prior section).[4]
The flow chart in Fig. 7 shows the iterative process (CANON-Algorithm) of how to uniquely label 2-(acetyloxy)benzoic acid. After the iterative process is finished all atoms are supposed to contain a number mathematically deduced from the molecular structure and therefore unique. For this purpose, invariant structural properties need to be introduced. Those are called invariant since they do not depend on the atom arrangement within the molecule.
Generally, every atomic property that fulfils this condition can be used and in this case the following invariant properties or ‘five atomic invariants’ are chosen:
(1) the number of connections
(2) the number of non-hydrogen bonds
(3) the atomic number
(4) the sign of the charge
(5) the number of attached hydrogens
The five atomic invariants are listed and numbered in order of a pre-defined prioritization.[4] The individual steps that are shown in Fig. 7 are explained step-by-step in the following paragraph. The headlines will contain the step number and the position within the figure in parenthesis.
Step Zero (first structure, first row):
- By applying the atomic invariants five integers are assigned to every atom of the molecule.
- The so-called individual invariants are obtained by simply combining these integers in the aforementioned order.
- E.g.: the methyl carbon of 2-(acetyloxy)benzoic acid in Fig. 7 has the individual invariant 110603.
- From this individual invariant, 1 connection, 1 bond to a non-hydrogen neighbour, an atomic number of 06, 0 charge and 3 attached hydrogen atoms can be concluded.
First Step (second structure, first row):
- After assigning every atom an individual variant, those are compared among all constituting (heavy) atoms and are ranked by magnitude to obtain the atomic rank.
- Next, The corresponding prime is assigned for every atomic rank (for 1, 2, 3, 4 the corresponding primes are 2, 3, 5, 7 and so on).
Second Step (third structure, first row):
- The so-called new invariant for a certain node (atom) is obtained by multiplying the corresponding primes of all neighbours of that node.
- That way the topological information around that node is incorporated into the labeling process.
- At this point, both the atomic rank and the new invariant are available for every non-hydrogen atom.
Third Step (fourth structure, first row):
- Afterwards, new atomic ranks are assigned, based on their current atomic rank and new invariant.
- The current atomic rank has a higher priority, hence the new invariant is only used to break ties between nodes that share the same atomic rank.
Fourth step (complete second row):
- The iterative process can be repeated with the new atomic ranks starting from the First step:
→calculate corresponding primes of the ranks
→calculate new invariants
→re-rank the nodes.

The process is over once the ranks are not changing anymore (if they did not change once, neither will they change in the following iterations).
If there are constitutionally symmetric nodes present in the molecular graph, it becomes necessary to break ties since the symmetric groups make it impossible to find a complete ranking. For tie-breaking, all ranks are doubled, and the first instance of a symmetric node is decremented by one. The resulting node ranking is considered a new invariant set that goes through the aforementioned iterative process (see Fig. 7) until it is no longer changing.
THE GENES part of the algorithm utilizes the uniquely ordered ranking to chose the starting node and prioritize at branching points, etc. As an entry point for the generation of canonical SMILES, the node with the lowest ranking is chosen. Branching decisions are made in the same fashion, i.e. the branching option with the lowest rank is chosen and followed until a dead end has been reached.
A special rule applies when branching into a ring with a double or triple bond. To avoid opening the ring at a multi-bond, the algorithm will always branch towards the multi-bond. Also, the ring-opening digits must be in the order of ring-opening nodes.
Conclusively, a unique SMILES string can be assigned by first generating a unique invariant rank for every node that incorporates invariant atomic properties and topological information and then using these ranks as decision indicators for branching, cycles and so on.[4]
Aftermath and Sources
Disclaimer: I generated the pictures used in this article by myself using the following open source software: Inkscape and Chemsketch.
[1] Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules Journal of Chemical Information and Modeling 1988, 28, 31–36.
[2] PowerPoint Presentation (bigchem.eu) by Martin Vogt, University of Bonn
[3] Daylight; last access 14.05.2021
[4] Weininger, D. et. al SMILES. 2. Algorithm for generation of unique SMILES notation Journal of Chemical Information and Modeling 1989, 29, 97–101
